| Text |
A
randomised trial without an intervention is difficult to envisage. In
today's Lancet, however, Nic Timpson and colleagues1 take advantage of
natural genetic randomisation during sexual reproduction-that of alleles
bearing single nucleotide polymorphisms (SNPs) governing the abundance
of C-reactive protein (CRP)-to assess whether associations between CRP
and components of the metabolic syndrome are causal.
In prospective observational studies, CRP concentration has consistently
been linked with cardiovascular events2 as well as with high-risk vascular
phenotypes including high blood pressure, diabetes, and the metabolic
syndrome.3,4 This fits with the prevailing view that inflammation is critical
to atherogenesis. Whilst the similar and well-known associations of blood
pressure and cholesterol levels with cardiovascular events are considered
causal-because reducing blood pressure or cholesterol reduces cardiovascular
risk in randomised trials-the same might not be true for CRP. Associations
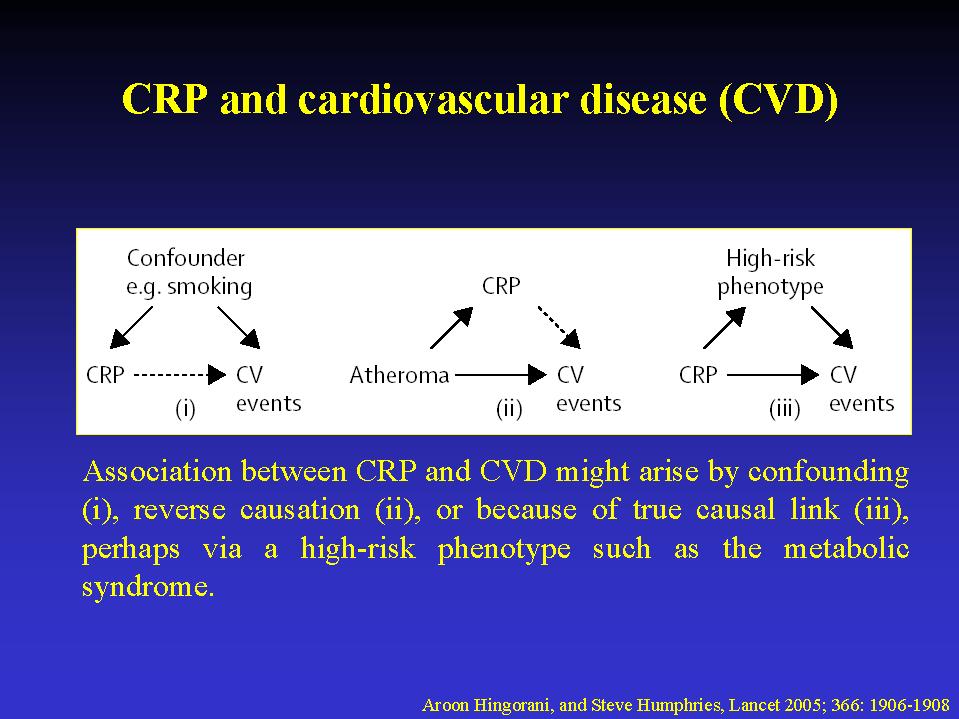
between CRP and disease could be explained by confounding, because of
CRP's associations with other risk factors such as low birthweight, lower
sociodemographic position, lack of physical activity, smoking, and abdominal
obesity.5,6 Reverse causation might also be at work, whereby inflammatory
cytokines from atheroma or adipose tissue raise CRP. (figure 1). While
statistical adjustment can reduce confounding, not all confounding factors
are known, or accurately measured. Moreover, adjustment requires a judgment
about mechanism. For example, if blood pressure or diabetes mediate rather
than confound the association between CRP and cardiovascular events, adjusting
for these factors would lead to underestimation of the causal association.
How might we get better insight into causation? Mechanistic studies in
vitro have yielded conflicting results. Potentially proatherogenic and
blood pressure-raising effects of CRP on vascular cells and tissues7 might
have been mediated by proinflammatory bacterial peptides or sodium azide
present in commercial CRP preparations.8,9 The increased atheroma formation
in apolipoprotein-E-deficient mice that overexpress human CRP was not
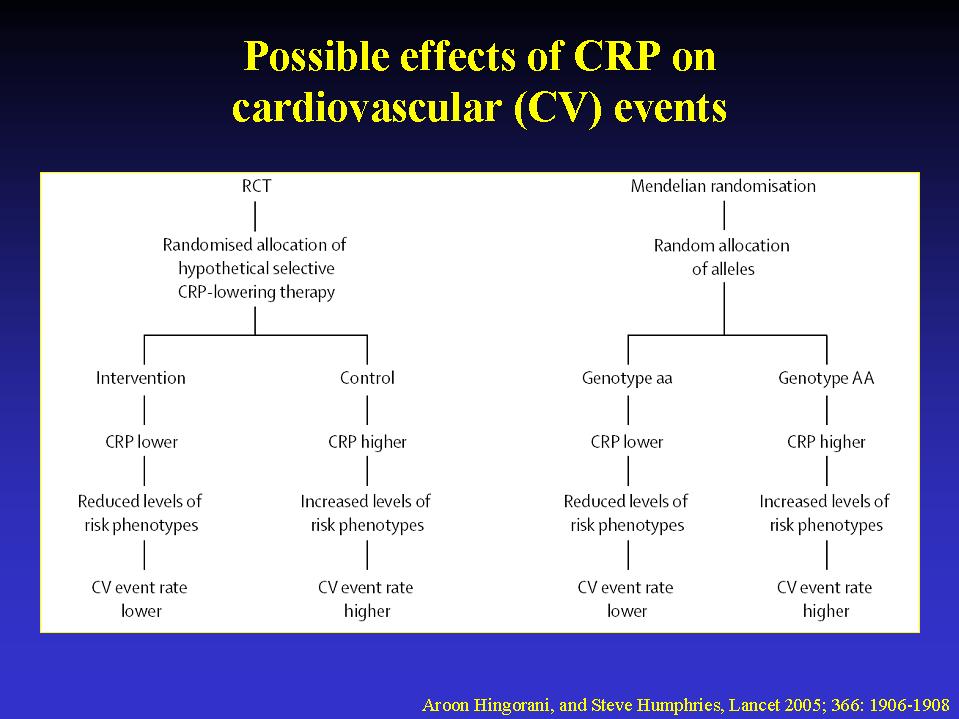
reproducible.10 A randomised trial of a selective CRP-lowering therapy
is required, because randomisation would ensure that measured and unmeasured
confounders were evenly distributed between placebo and intervention groups
(figure 2). Unfortunately, no selective CRP-lowering drug exists. Statins
lower CRP and have beneficial effects on cardiovascular disease, but the
benefits may be adequately explained by cholesterol lowering. Alleles
of the gene encoding CRP exist that influence circulating CRP concentration.
These are transmitted from parent to offspring at random. Therefore, factors
that could confound associations of CRP with components of the metabolic
syndrome should be distributed evenly in those who do, and those who do
not, have high-CRP alleles (figure 2). Moreover, because genotype is determined
before onset of disease, the possibility of reverse causation is also
overcome.11,12 Timpson and coworkers confirm that CRP is associated with
metabolic syndrome, and that SNPs and haplotypes of the CRP gene are strongly
associated with differences in CRP concentration. As expected, haplotypes
inferred from these SNPs were not associated with factors, such as smoking,
that could confound the association of CRP with the metabolic syndrome.
By quantifying the associations of CRP haplotypes with blood pressure,
triglycerides, HDL-cholesterol, adiposity, and insulin resistance, Timpson
and colleagues made indirect but unconfounded estimates of the association
of CRP with these components of the metabolic syndrome. In all cases,
the indirect genetic estimates were smaller than the directly observed
associations of CRP with the same factors, and in some cases they were
in the opposite direction. This analysis suggests that the directly observed
associations of CRP with the metabolic syndrome are affected by residual
confounding and/or reverse causation, leading to the conclusion that CRP
is unlikely to be a causal factor in its development. These findings could
have wider implications. First, the data suggest that components of the
metabolic synd-rome probably act as confounders, and do not mediate the
association between CRP and cardiovascular events. Thus, it is probably
legitimate to adjust for these factors when examining associations of
CRP with cardiovascular events in observational studies. Second, the study
identifies a set of genetic tools for examining whether CRP associations
with cardiovascular events are causal. This is valuable because it would
provide a test for claims that CRP-lowering could be as important as cholesterol-lowering
in the prevention of cardiovascular events. How robust are Timpson and
colleagues' conclusions? First, it is assumed that CRP genotype acts only
by altering the circulating concentration of CRP but not its function,
an assumption that appears reasonable with current evidence. Second, it
assumes that a lifelong, genetically-determined increase in CRP does not
lead to compensatory changes in other systems to inhibit potentially adverse
effects of a higher CRP level.11 This could lead to the erroneous conclusion
that raised CRP in later life as a result of adverse behaviours is not
important in the development of the metabolic syndrome or cardiovascular
disease. Finally, because differences in CRP level by genotype are small,
very large studies will be required to reduce the present imprecision
of the unconfounded genetic estimates of the effect of CRP on the metabolic
syndrome. Even larger studies will be needed to assess whether or not
associations of CRP with cardiovascular events are causal, and it is likely
that studies will need to be pooled to achieve adequate sample size. Nevertheless,
Mendelian randomisation offers the potential for insight into causation
beyond that usually possible from observational epidemiology.11 Many other
circulating biomarkers are also associated with cardio-vascular disease
risk but, as with CRP, specific drugs that lower their concentration do
not exist, and there are uncertainties as to whether such associations
are causal. If it were possible to distinguish probable causal from non-causal
links using Mendelian randomisation, this might help prioritise the development
of new drugs for cardiovascular disease prevention. With the recent publication
of databases of human DNA sequence variation,13 the availability of genetic
tools for Mendelian randomisation studies is increasing. Carefully phenotyped
cohorts, such as Timpson and colleagues', will facilitate such analyses
and are testimony to the importance of a strong interface between genetics
and epidemiology.
References
1) Timpson NJ, Lawler DA, Harbord RM, et al. C-reactive
protein and its role in metabolic syndrome: mendelian randomisation study.
Lancet 2005; 366: 1954-59.
2) Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and
other circulating markers of infiammation in the prediction of coronary
heart disease. N Engl J Med 2004; 350: 1387-97.
3) Pradhan AD, Cook NR, Buring JE, Manson JE, Ridker PM. C-reactive protein
is independently associated with fasting insulin in nondiabetic women.
Arterioscler Thromb Vasc Biol 2003; 23: 650-55.
4) Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive
protein and the risk of developing hypertension. JAMA 2003; 290: 2945-51.
5) Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J
Clin Invest 2003; 111: 1805-12.
6) Sattar N, McConnachie A, O'Reilly D, et al. Inverse association between
birth weight and C-reactive protein concentrations in the MIDSPAN Family
Study. Arterioscler Thromb Vasc Biol 2004; 24: 583-87.
7) Verma S, Wang CH, Li SH, et al. A self-fulfilling prophecy: C-reactive
protein attenuates nitric oxide production and inhibits angiogenesis.
Circulation 2002; 106: 913-19.
8) Taylor KE, Giddings JC, van den Berg CW. C-reactive protein-induced
in vitro endothelial cell activation is an artefact caused by azide and
lipopolysaccharide. Arterioscler Thromb Vasc Biol 2005; 25: 1225-30.
9) Pepys MB. CRP or not CRP? That is the question. Arterioscler Thromb
Vasc Biol 2005; 25: 1091-94.
10) Hirschfield GM, Gallimore JR, Kahan MC, et al. Transgenic human C-reactive
protein is not proatherogenic in apolipoprotein E-deficient mice. Proc
Natl Acad Sci USA 2005; 102: 8309-14.
11) Davey Smith G, Ebrahim S. 'Mendelian randomization': can genetic epidemiology
contribute to understanding environmental determinants of disease? Int
J Epidemiol 2003; 32: 1-22.
12) Keavney B. Commentary: Katan's remarkable foresight: genes and causality
18 years on. Int J Epidemiol 2004; 33: 11-14.
13) Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes
with linkage disequilibrium. Lancet 2005; 366: 1223-34.
|