| Author | Heribert Schunkert, and Nilesh J. Samani. | |||
| Title | Elevated C-Reactive Protein in Atherosclerosis - Chicken or Egg? | |||
| Full source | N Engl J Med 2008;359:1953-1955 | |||
| Text |
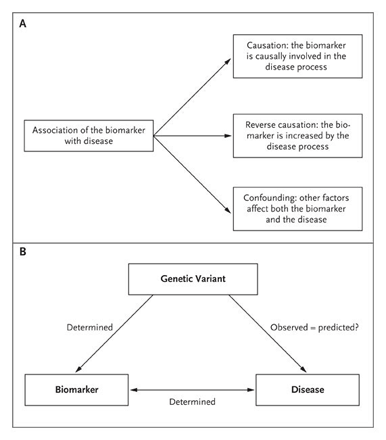
One of the most debated topics in cardiovascular medicine is whether C-reactive protein (CRP), a component of the acute-phase response, is a causal factor in the pathogenesis of atherosclerosis.1,2 If it is, the implications could be far-reaching and include new approaches for the prevention and treatment of myocardial infarction and stroke.3 Support for a role of CRP in the pathogenesis of atherosclerosis comes largely from epidemiologic studies that have consistently observed an association between elevated plasma CRP levels and cardiovascular events.1,4,5 The statistical strength of such associations is at least as robust as that of established risk factors such as hypertension, diabetes mellitus, and hypercholesterolemia.5 However, statistical strength does not imply causality, since confounding factors or reverse causality offer alternative explanations for the association (Figure 1A). CRP is particularly susceptible to confounding, since multiple cardiovascular risk factors, including smoking, hypertension, obesity, lack of physical activity, and low socioeconomic status, all relate independently to elevated plasma levels of the protein.7,8 Reverse causation is also a potential explanation, since atherosclerosis may trigger an elevation of CRP levels.
For established cardiovascular risk factors, causality was proved by randomized treatment trials showing the clinical benefits of lowering blood pressure, cholesterol levels, and glucose levels. As reported in this issue of the Journal, Zacho and coworkers6 used a less familiar study design for testing the causality of CRP in atherosclerosis. The instrument these investigators used is genetic variation in the CRP gene that gives rise to variation in plasma CRP levels. They took advantage of the fact that persons are effectively randomly assigned at birth to either higher or lower CRP levels depending on the genetic variant they receive from their parents.9 According to Mendel's law of independent assortment, neither endogenous nor exogenous factors disturb this randomization process, which therefore can be termed "mendelian randomization."9 (This generalization is not entirely free of exceptions, since genes that are very close to one another on a chromosome do not segregate independently; however, it is probably true for most traits.) Zacho and coworkers investigated the three component associations of a mendelian randomization study (Figure 1B). First, the effect of CRP genotypes on plasma CRP levels was quantified (Figure 1B, left line of the triangle). This is relatively easy and can be done with great precision. Second, the magnitude of the association between plasma CRP levels and ischemic events (but not the causality of the association) was quantified (Figure 1B, bottom line of the triangle). Finally, using the information from these two analyses, Zacho et al. predicted the expected effect of the genetic variants on ischemic events (Figure 1B, right line of the triangle). If the predicted effect and the actually observed effect agreed, one could infer that the relationship between plasma CRP level and the risk of atherosclerotic disease was causal. The most crucial part in such a study is to obtain a sample size that allows precise estimations of the effect sizes. Zacho et al. measured high-sensitivity CRP levels and conducted genotyping for four CRP genetic variants in 50,816 subjects. They found that genetic CRP variants explained a difference in plasma CRP levels of up to 64% (Figure 1B, left side). Moreover, the investigators observed the expected association between the plasma CRP level and cardiovascular disease (Figure 1B, bottom). Despite confirming these two associations, the authors found that none of the CRP variants were associated individually or in combination with the risk of ischemic events (Figure 1B, right side). Crucially, Zacho et al. were able to reliably estimate the effect on ischemic events that the genetic variants should have had, and they showed that the observed association was markedly different from the predicted effect. As further proof of the validity of their approach, Zacho et al. included a positive control in their experiment: they typed variants in the apolipoprotein E gene, which affect cholesterol level, and found the predicted association between these variants and increased cardiovascular risk. A positive feature of the study by Zacho et al. is that the various associations were examined in the same population, rather than extrapolated from individual associations observed in different cohorts. A limitation of the study is the partial use of cross-sectional and especially case–control cohorts in the analysis, since the use of subjects recruited after the event could introduce a survival bias. The proposed interpretation of the findings relies on the assumption that genetically elevated CRP levels behave similarly to acquired elevations in CRP levels (due to inflammation, for example). It also assumes that lifelong genetic elevation of plasma CRP levels does not induce compensatory mechanisms in other systems. Nevertheless, the findings are conclusive and consistent with other mendelian randomization studies of CRP.10,11,12,13 What are the implications of the findings? First, they strongly indicate that CRP is not causally involved in the pathogenesis of atherosclerotic disease. Thus, immediate targeting of CRP is unlikely to be beneficial in reducing the risk of cardiovascular events. For definitive proof, this hypothesis needs to be tested in randomized clinical trials of CRP inhibitors, but the findings of Zacho et al. and other investigators who have performed genetic studies of CRP10,11,12,13 argue against a positive outcome of such studies. However, the findings should not be interpreted as suggesting that factors that cause acquired elevations in CRP levels, such as inflammation, do not play a causal role in atherosclerosis. In fact, the data suggest that confounding factors not considered by the multivariate analysis (such as inflammation) affect the regulation of CRP and the risk of ischemic disease in parallel. Moreover, the study does not diminish the validity of CRP as a risk marker of atherosclerotic disease. Clinical studies such as the recently terminated Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin (known as JUPITER, ClinicalTrials.gov number, NCT00239681 [ClinicalTrials.gov] )14 will establish whether measuring CRP is useful for risk stratification and therapeutic decision making. From a broader perspective, the study by Zacho et al. provides a landmark example of how genetics may help to illuminate research in the cardiovascular field. Indeed, modern genomic analysis not only may identify new risk genes and thereby mechanisms leading to coronary artery disease15 but also may allow the functionality of circulating risk markers to be determined. No
potential conflict of interest relevant to this article was reported.
References
|
|||